

-61970c89f24d853d.jpg)
Reportagem: Camila Neumam
Fotos: José Felipe Batista
Um estudo realizado por pesquisadores do Centro de Vigilância Viral e Avaliação Sorológica (CeVIVAS) do Instituto Butantan, em conjunto com outras 23 instituições, incluindo a Universidade Yale, dos Estados Unidos, define um novo sistema de nomenclatura para linhagens do vírus da dengue (DENV) a fim de complementar a classificação por genótipo.
O objetivo é facilitar a vigilância genômica, ou seja, favorecer a comunicação entre laboratórios e autoridades de saúde, tornando o rastreamento de linhagens mais preciso e comparável, além de acompanhar potenciais linhagens com relevância epidemiológica ou clínica.
A pesquisa A new lineage nomenclature to aid genomic surveillance of dengue vírus (Uma nova nomenclatura de linhagem para auxiliar na vigilância genômica do vírus da dengue, em tradução livre) foi publicado na revista científica PLOS Biology.
.jpg)
Os bioinformatas Alex Ranieri e James Siqueira, do CeVIVAS, desenvolveram, em conjunto com 23 institutições, um novo sistema de nomeclatura para linhagens do vírus da dengue
“Pelo ponto de vista epidemiológico, a dengue que circula é a mesma dos últimos 20 anos, e parece que o vírus não está variando, quando na verdade está. A nova nomenclatura faz essa classificação de forma sistemática, permitindo, além da facilidade da identificação, a qualquer pesquisador classificar essas amostras”, afirma o bioinformata do CeVIVAS e do Laboratório de Ciclo Celular do Butantan (LCC), Alex Ranieri, um dos principais autores do estudo.
A dengue é uma doença viral transmitida pelo mosquito Aedes aegypti, que coloca em risco mais de 100 milhões de pessoas por ano no mundo, segundo a Organização Mundial da Saúde (OMS), especialmente em países tropicais como o Brasil.
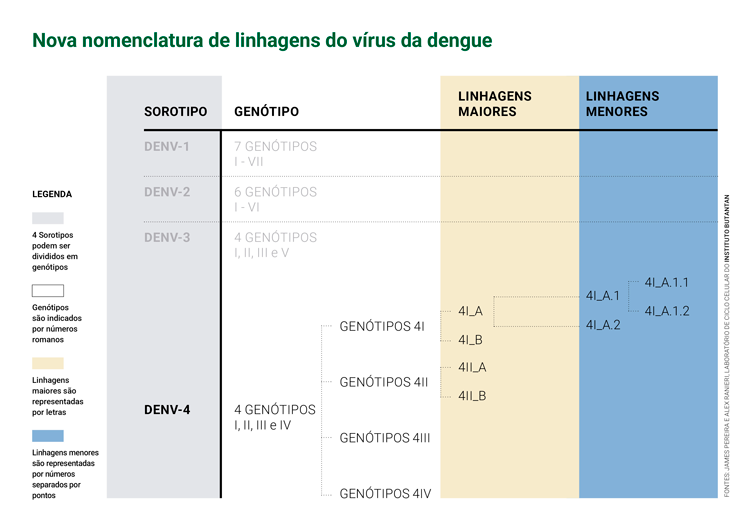
Até hoje, foram identificados quatro sorotipos do vírus da dengue – DENV-1, DENV-2, DENV-3 e DENV-4. Dentro de cada sorotipo existe diferentes variações genéticas, chamadas genótipos. Com o tempo, esses vírus acumulam mutações em seu material genético (RNA) e dão origem a linhagens, que representam ramos mais recentes ou mais antigos da história evolutiva do vírus.
Em outras palavras: primeiro distinguimos os vírus da dengue pelo sorotipo, que podem ser divididos em diferentes genótipos. No novo esquema de nomenclatura do vírus da dengue, os genótipos são indicados por números romanos (ex.: genótipo III), e dentro deles existem dois níveis hierárquicos: as linhagens maiores representadas por letras (ex.: DENV-3III_C), e as linhagens menores indicadas por números separados por pontos (ex.: DENV-3III_C.2). Esse sistema pode ser expandido conforme necessário, gerando sublinhagens como DENV-3III_C.2.1, de modo semelhante ao esquema usado para o SARS-CoV-2 (vírus causador da Covid-19). Assim, cada nome carrega informação evolutiva e epidemiológica, permitindo identificar com clareza a relação entre diferentes grupos virais.
“Não havia uma forma padronizada de classificar o que estava abaixo do genótipo, dificultando tanto a análise da variedade do vírus nessa camada, quanto a comunicação científica e epidemiológica com as autoridades de saúde pública”, afirma o bioinformata James Siqueira Pereira, aluno de doutorado vinculado a Escola Superior do Instituto Butantan (ESIB) e ao Programa Interunidades de Bioinformática da Universidade de São Paulo (USP) sob orientação de Alex Ranieri.
Em 2024, os países das Américas, onde circulam os quatro sorotipos de dengue, notificaram mais de 13 milhões de casos; destes, quase 7 milhões foram confirmados em laboratório. O Brasil foi o país com o maior número de casos (10,2 milhões); seguido por Argentina (581,5 mil); México (558,8 mil); Colômbia (321 mil); e Paraguai (295,7 mil), segundo a Organização Pan-Americana da Saúde (OPAS).
.jpg)
A nomenclatura das linhagens foi definida levando em consideração os princípios da Phylogenetic Assignment of Named Global Outbreak Lineages (Pango lineages), sistema hierárquico criado para rastrear a evolução das variantes do SARS-CoV-2; bem como a lógica de sistemas mais antigos, como o que é utilizado para nomear os vírus influenza.
A metodologia da pesquisa partiu da análise de uma vasta base de sequências genômicas de DENV, muitas das quais geradas pelo projeto CeVIVAS. As linhagens foram definidas com limiares altos de distância filogenética (ramificações que apontam diferentes mutações do vírus) [VEJA O INFOGRÁFICO] e tamanho do clado, que são os grupos de sequências que compartilham um ancestral comum e seus descendentes.
O sistema foi implementado em ferramentas de atribuição de linhagens como Genome Detective, GLUE e NextClade, capazes de realizar a classificação mesmo com sequências parciais.
“Ao tornar o sistema compatível com as classificações existentes e demonstrar sua utilidade, pretendemos alcançar ampla aceitação e introduzir uma linguagem global verdadeiramente padronizada para discutir a diversidade genética do DENV”, afirma James Siqueira.
A aplicação do sistema foi feita com amostras de vírus dengue circulantes em diversas localidades geográficas demonstrando o rastreamento temporal de linhagens dos quatro sorotipos em um contexto global.
“O NextClade, uma das ferramentas onde o sistema foi implementado é capaz de analisar de forma fácil e rápida um grande número sequências, podendo ser utilizado por pessoas com e sem experiência em bioinformática. Além disso, para manter tudo dentro das normas, há um comitê que se reúne e se debruça sobre as sequências, avalia se há divergências, e monitora linhagens com grande possibilidade de serem novas”, informa James Siqueira.
Por possibilitar a classificação de linhagens mesmo com cobertura genômica parcial, o sistema se torna uma ferramenta prática em cenários com dados limitados. Além disso, também se mostrou útil para desenvolvimento de vacinas, estudos de transmissão e potencial virulência.
“Este trabalho apresenta uma proposta robusta para padronizar o rastreamento de linhagens do vírus da dengue, com ferramentas públicas de precisão, mesmo com dados incompletos. A nomenclatura hierárquica de linhagens (maior/menor) promete aprimorar a vigilância global do vírus da dengue”, ressalta Alex Ranieri.
Segundo os pesquisadores, a nova classificação ganha importância diante da expansão da dengue no Brasil e no mundo, sobretudo em áreas não endêmicas, como nos Estados Unidos e na Europa, ao mesmo tempo que a capacidade global de sequenciamento também se expande a partir da integração aos sistemas de saúde pública.
“A ideia é facilitar e fornecer as ferramentas para quem é da epidemiologia e para quem toma decisões de saúde pública, para que tenham uma forma sistemática de definir o que está circulando agora, comparar com o que estava circulando anteriormente, e as implicações disso”, esclarece Alex Ranieri.
.jpg)
Os quatro sorotipos do DENV e seus genótipos foram identificados no final da década de 1990 e início dos anos 2000 com base em sequências genéticas parciais.
Apesar de todos causarem a mesma doença, a infecção por um dos quatro sorotipos reserva imunidade apenas para aquele vírus específico, não para os três outros sorotipos. Ao contrário, infecções subsequentes por outros sorotipos aumentam a chance de formas mais severas da doença, como a dengue grave, que por muito tempo ficou conhecida como dengue hemorrágica.
Segundo o Ministério da Saúde, a primeira epidemia de dengue documentada clínica e laboratorialmente no Brasil ocorreu no começo da década de 1980, em Boa Vista (RR), causada pelos sorotipos 1 e 4. Em 1986, ocorreram epidemias no estado do Rio de Janeiro e em algumas capitais do Nordeste.
Desde então, a ocorrência da forma endêmica da doença se intercala com anos de epidemias, geralmente associadas à introdução de novos sorotipos em áreas sem transmissão e/ou alteração do sorotipo predominante, acompanhando a expansão do mosquito vetor.
Segundo o Painel de Monitoramento de Arboviroses do Ministério da Saúde, o aumento da incidência da dengue no Brasil entre 2000 e 2002 foi associado à introdução do DENV-3, elevando o risco de epidemias de dengue e febre hemorrágica da dengue. Em 2024, o país viveu um grande aumento nos números da doença provocado pela reintrodução desse sorotipo, com a notificação de mais de 6,5 milhões de casos prováveis e mais de 6.300 mortes.
“A evolução e disseminação contínuas do vírus da dengue levaram ao surgimento de linhagens evolutivas distintas dentro de genótipos reconhecidos. Além disso, com a implementação de intervenções, como as vacinas e os mosquitos infectados que podem eventualmente selecionar linhagens virais específicas, é imperativo ter uma linguagem precisa e comum para monitorar a transmissão contínua do dengue em diferentes escalas espaço-temporais, e que isso seja comunicável a médicos e autoridades de saúde pública que podem não ter experiência em genômica”, conclui Alex Ranieri.



